
Pendant des décennies, les chercheurs ont cherché à éclaircir les causes des troubles neurodégénératifs, un groupe de maladies dévastatrices, notamment la maladie d'Alzheimer et la maladie de Parkinson, qui impliquent la perte progressive des neurones et le dysfonctionnement du système nerveux. Si la compréhension précise des liens entre le vieillissement et la neurodégénérescence reste encore un défi, ces recherches de la faculté de médecine de Harvard apportent de nouveaux et précieux indices. Mutations génétiques liées à l’âge, infections virales, alors que de nombreux facteurs sont soupçonnés dans ce processus, l’équipe de recherche décrit, dans la revue Cell, sa découverte d’un lien moléculaire entre le vieillissement et une cause génétique majeure de la sclérose latérale amyotrophique (SLA) et de la démence fronto-temporale (FTD), 2 maladies neurodégénératives.
Des résultats qui désignent de nouvelles cibles possibles pour le traitement de ces maladies et d’autres maladies neurodégénératives. Car l’étude fournit la première description d'un événement moléculaire qui relie le vieillissement à la neurodégénérescence, explique l'auteur principal de l'étude, Junying Yuan, professeur de biologie cellulaire à Harvard. « Des connaissances qui constituent une étape critique dans la compréhension des mécanismes par lesquels le vieillissement prédispose les individus à la neurodégénérescence ». Des résultats également, qui soulignent la nécessité d'une meilleure compréhension de la biologie des maladies neurodégénératives dans le contexte du vieillissement.
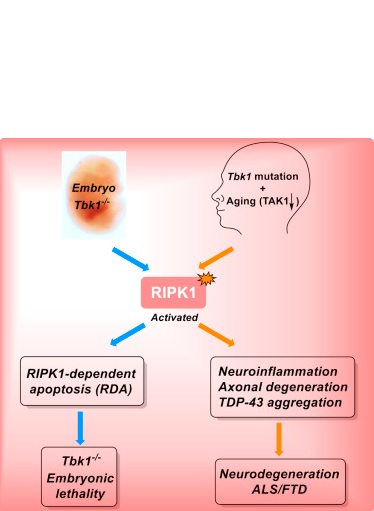
Une protéine déjà impliquée dans une forme de mort cellulaire programmée : la SLA (sclérose latérale amyotrophique ou maladie de Lou Gehrig) est caractérisée par la mort progressive des motoneurones. La SLA partage certaines caractéristiques cliniques et génétiques avec la démence fronto-temporale caractérisée par une démence précoce et rapide. Environ un patient sur dix présentant les deux maladies est porteur de mutations génétiques provoquant un dysfonctionnement partiel d’une protéine appelée TBK1. Des niveaux réduits de TBK1 sont trouvés chez les patients atteints de SLA et de DFT. De plus, de précédentes études ont montré que la protéine TBK1 est impliquée dans une forme de mort cellulaire programmée et dans la neuroinflammation, caractéristique des troubles neurodégénératifs. La manière dont TBK1 contribue au développement de la SLA et du DFT restait cependant mal comprise.
TBK1 et TAK1, 2 protéines clés qui se réduisent avec le vieillissement : l’équipe montre ici que des souris modèles conçues pour avoir des niveaux réduits de TBK1 ne peuvent vivre en bonne santé qu’à condition de bloquer l'activité de RIPK1, une autre protéine jouant un rôle central dans la mort cellulaire programmée, la neuroinflammation et les maladies neurodégénératives. Des analyses supplémentaires ont révélé que TBK1 inhibe l'activité de RIPK1 pendant le développement embryonnaire. Cette découverte a incité les chercheurs à étudier une autre protéine, appelée TAK1, connue auparavant pour inhiber également la fonction RIPK1. Lorsqu'ils ont examiné les données sur l'expression de TAK1 dans le cerveau humain, les scientifiques ont constaté que l'expression de TAK1 diminuait de manière significative avec l'âge. Dans les cerveaux des patients atteints de SLA, l'expression de TAK1 était encore réduite par rapport au cerveau de personnes du même âge sans SLA.
Pour modéliser l'interaction entre la perte partielle de TBK1 et TAK1 avec le vieillissement, l'équipe a créé des souris exprimant la moitié de la quantité habituelle de TBK1. Les souris ont également exprimé la moitié de la quantité habituelle de TAK1 dans leur microglie, les cellules immunitaires du cerveau où TAK1 est normalement le plus actif.
Des réductions à la fois de TBK1 et de TAK1 induisent des traits associés à la SLA et à la DFT, montre ensuite l’équipe toujours sur la souris, notamment des déficits moteurs, une faiblesse des membres postérieurs, un comportement anxieux dans de nouveaux environnements et des modifications de la chimie cérébrale. Les souris présentaient une réduction du nombre de neurones dans le cerveau et une augmentation du dysfonctionnement des neurones moteurs et de la mort cellulaire. Mais lorsque l’équipe inhibe l’activité de RIPK1 indépendamment de TBK1 et TAK1, elle observe une inversion des symptômes.
TAK1 et TBK1 travaillent ensemble pour supprimer l'activité délétère de RIPK1, et si l’une « tombe en panne », l'autre peut compenser. Mais si les deux protéines sont réduites, l'activité de RIPK1 augmente, entraînant la mort cellulaire et la neuroinflammation.
De nouveaux traitements en vue ? Des essais cliniques sont en cours pour tester l'innocuité et l'efficacité de médicaments bloquant l'activité de RIPK1 dans les maladies inflammatoires neurodégénératives et chroniques. Ces résultats confirment la justification de ces essais, commentent les chercheurs, qui suggèrent que dans les 2 ans, les inhibiteurs de RIPK1 pourraient se révéler efficaces dans le traitement de SLA et de la DFT.
Un immense espoir alors qu’aucun traitement efficace n'a encore été mis au point pour les maladies neurodégénératives et, qu’avec le vieillissement des populations, leur prévalence explose.
Source : Cell August 23, 2018 DOI: 10.1016/j.cell.2018.07.041 TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging
Plus sur Neuro Blog